Science:基于机器学习准确预测偶联反应收率
更新日期:2018-05-09在药物研发和天然产物的合成中,化学家不仅要设计出合理的合成路线还要尽可能的提高每一步反应的产率,显然利用传统的有机合成方式进行这一工作,不仅是对成本(时间、金钱)和实验工作者体力的挑战,也是对路线设计者脑力(记忆力和分析能力)的挑战。然而近年来智能化和自动化技术的迅猛发展正让这类化学合成的传统实验室工作模式发生着翻天覆地的变化。即便如此,现有的反应自动优化模式似乎还无法满足合成化学的需求,能否“不战而屈人之兵”,即,不做实验仅通过计算来预测反应收率?
近日,来自普林斯顿大学的Abigail Doyle教授与美国默克的Spencer Dreher博士等研究人员合作使用了一种强大的机器学习算法——随机森林算法(random forest algorithm),在接受数以千计的Buchwald-Hartwig偶联反应数据的训练后,这种算法可以准确预测其他具有多维变量的Buchwald-Hartwig偶联反应收率。就算训练数据大幅减少到数百个反应,或者预测样本外反应,这种算法的表现依然十分亮眼。相关进展“Predicting reaction performance in C–N cross-coupling using machine learning”发表在近期的Science杂志上(Science, 2018, DOI: 10.1126/science.aar5169)。
反应模型的选择
异噁唑及其衍生物是一类含N-O键的五元杂环化合物,该结构广泛存在于生物活性分子及天然产物中,并且具有很好的药理学特性。Buchwald-Hartwig偶联反应是药物合成中应用最为广泛的反应之一,然而使用以异噁唑结构为官能团的化合物进行Buchwald-Hartwig偶联反应构建复杂药物分子依然面临着巨大的挑战。借鉴德国明斯特大学Frank Glorius教授的方法,作者以异噁唑化合物为添加剂时的甲基苯胺与卤代芳烃的Buchwald-Hartwig偶联反应为模板开始了他们的人工智能(AI)之旅。
反应数据的获得
正所谓“种瓜得瓜种豆得豆”,很多机器学习算法的效果关键在于训练数据的质量。在化学领域,机器学习的数据主要来源于那些已经发表过的文献或者专利中的成功反应。Abigail Doyle教授表示,这项研究之所以能取得成功的一个关键因素在于他们能够依靠美国默克开发的高通量反应筛选平台(点击阅读相关)。该平台让他们能在极短的时间内完成4608个Buchwald-Hartwig偶联反应,其中包括15种卤代芳烃和卤代杂环芳烃底物,23种异噁唑添加剂,4种钯配体和3种有机碱,反应溶剂为DMSO,反应温度为60 ℃,反应时间为16小时。这为机器学习提供了充足可靠的数据,包括非常重要的大量失败反应的数据。
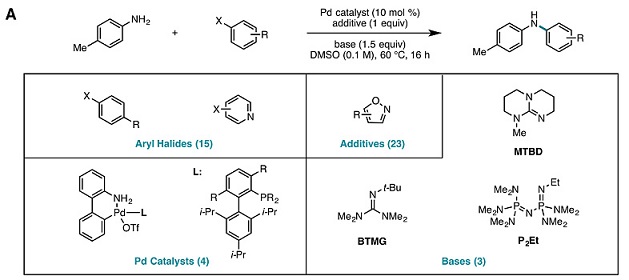
图1. 以含四个变量的Buchwald-Hartwig偶联为模板通过自动反应平台筛选收集反应数据。图片来源:Science
样本集的生成
描述符是机器学习的基础,精确定义且合理选择与研究对象相关的描述符在机器学习中十分重要。作者将反应中四种变量的化合物结构分类并写入Spartan软件中,随后通过计算挑选出与体系相关的120种描述符,包括分子描述符:轨道能量(EHOMO和ELUMO)、偶极矩、电负性、硬度、体积、质量、椭圆度及表面积等;原子描述符:原子静电荷和核磁位移等;振动描述符:振动频率和强度等。其中用于描述异噁唑的参数有19个,描述卤化物的参数有27个,描述有机碱的参数有10个,而描述钯配体的参数则多达64个。如图3所示,以这些化合物的描述符为输入值,以反应产率为输出值,它们经转换为机器可分析的格式后构建起机器学习的样本集,其中矩阵的每一行为一个反应样本,每一列为样本的某一个特征。而机器学习的目的则是要通过样本集的训练建立起输入值与输出值间的映射关系,以便准确预测一个未知的新样本。
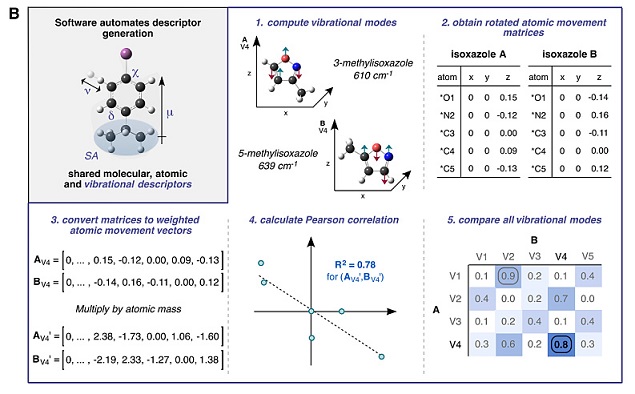
图2. 利用Spartan软件计算化合物的分子、原子和振动描述符。图片来源:Science

图3. 机器学习的样本集。图片来源:Science
机器学习及预测
作者从原始数据集中抽取70%的样本作为训练集用于训练预测算法,剩下的30%样本作为测试集用于检验预测模型的性能,其中在训练集中又采用K-交叉验证方式让机器学习样本,他们对多种线性回归模型(Linear regression models)、k-近邻算法(kNN)、支持向量机(SVM)、贝叶斯广义线性模型(Bayers GLM)、神经网络模型(Neutral network)和随机森林算法(Random forest algorithm)进行了考察。结果显示,接受训练后的各种算法中表现最优异的是随机森林算法,在对测试集的检验中,其预测产率值与实际产率值间的线性相关度最高,标准误差值(RMSE)为7.8%,同时R2值为0.96。通常机器学习的数据越少,其预测能力也逐渐下降,在对反应产率的预测上随机森林算法上也不例外。但是相比于其他模型需要用70%的数据进行训练才能达到一定的预测准确度而言,随机森林算法只需对5%的数据进行学习即可超越他们,这意味着要练就相同的“功力”,随机森林算法仅用230个反应即可,而其他算法却用了3225个反应。

图4. 经训练后的各种模型预测反应产率和实际反应产率的相关性分析。图片来源:Science
为了进一步展示这种随机森林算法的强大能力,他们又对另外8种异噁唑添加剂(16-23)参与的反应进行了预测,值的一提的是,这些新的添加剂此前并未在样本中出现并且其取代基与之前的15种添加剂(1-15)也不类似。结果表明,对于这些样本外的反应,随机森林算法同样能给出精准的预测,每种添加剂参与反应的产率预测值与真实值间的相关性分析如图5B所示,其平均RMSE值为11.3%,平均R2值为0.91。
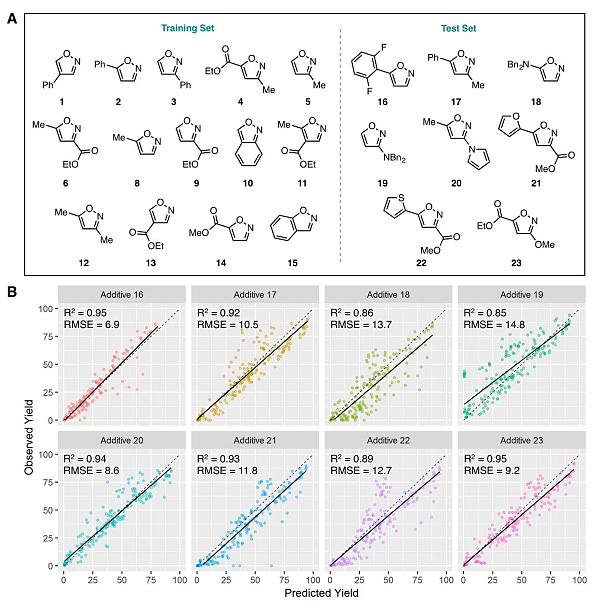
图5. 随机森林算法对样本外反应产率的预测值与实际值间的相关性分析。图片来源:Science
Abigail Doyle认为这项研究中AI能有如此神勇的表现主要归功于超大的高质量学习数据库的建立以及合理的化学描述符的选择,当然这也离不开高通量反应和化学计算软件Spartan的帮助。当然她也承认目前的方法仍然还有很多需要改进的地方,例如仅使用溴代芳香烃的反应数据对机器进行训练后,要预测氯代或碘代芳香烃参与反应的表现情况还有些困难。同时,由于反应原料和添加剂的剩余量无法准确测定,他们还无法通过该方法来间接评估含有官能团异噁唑底物的反应表现,这在一定程度上与他们最初的目标相背离。Doyle希望机器学习能够预测那些具有更复杂三维结构的底物参与的反应。
(来源:X-MOL)